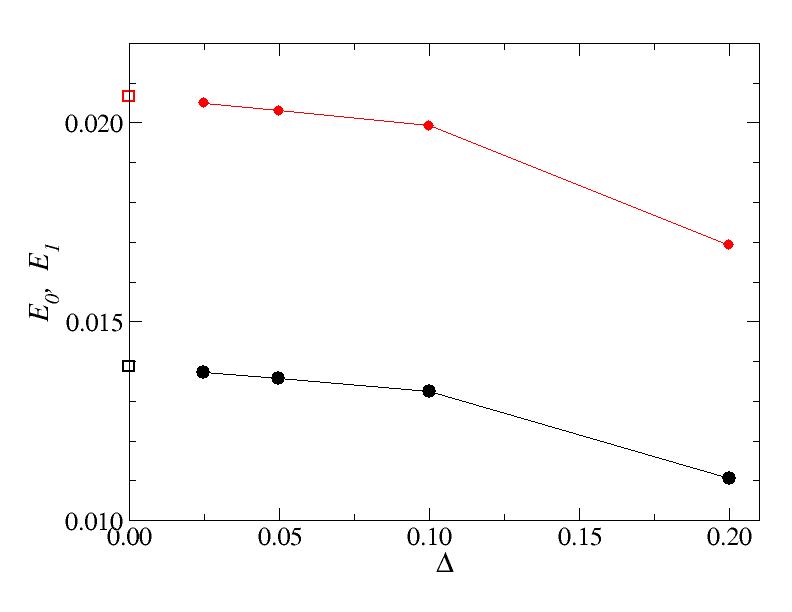
Problem A (variational calculation)
Solution program (instructor's Fortran version) [var.f90]
First, let's look at how the energies of the first four states converge as a function of the number of basis states, N=Nx*Ny, first using Nx=Ny:
Nx Ny Nx*Ny E0 E1 E2 E3 ------------------------------------------------------------------------ 5 5 25 0.01455951 0.02205329 0.04358191 0.06356153 10 10 100 0.01420417 0.02106122 0.04320811 0.06354212 15 15 225 0.01396114 0.02087201 0.04298447 0.06353202 20 20 400 0.01392182 0.02071306 0.04294863 0.06352973 25 25 625 0.01389049 0.02068858 0.04291843 0.06352809 30 30 900 0.01388495 0.02067539 0.04291384 0.06352769 35 35 1225 0.01388330 0.02067261 0.04291210 0.06352759 40 40 1600 0.01388304 0.02067227 0.04291190 0.06352755 45 45 2025 0.01388291 0.02067210 0.04291175 0.06352754 50 50 2500 0.01388262 0.02067129 0.04291153 0.06352751 55 55 3025 0.01388170 0.02067057 0.04291075 0.06352745 60 60 3600 0.01388129 0.02066912 0.04291042 0.06352741
Since the system is rectangular, and longer in the y-direction, one might expect the convergence to be faster for rectangular lattices with Ny > Nx. Let's look at results for Ny=2*Nx:
Nx Ny Nx*Ny E0 E1 E2 E3 ------------------------------------------------------------------------ 5 10 50 0.01422518 0.02110244 0.04322777 0.06355619 10 20 200 0.01392777 0.02072316 0.04295374 0.06353090 15 30 450 0.01388708 0.02067886 0.04291568 0.06352840 20 40 800 0.01388417 0.02067411 0.04291288 0.06352777 25 50 1200 0.01388315 0.02067215 0.04291198 0.06352767 30 60 1800 0.01388166 0.02066972 0.04291074 0.06352749 35 70 2400 0.01388077 0.02066861 0.04290998 0.06352742 40 80 3200 0.01388065 0.02066840 0.04290986 0.06352739
Indeed, for large N the energy here is lower (compare, e.g., the results for 45*45 and 30*60).
For completeness, let's also look at the case Ny=Nx/2:
Nx Ny Nx*Ny E0 E1 E2 E3 ------------------------------------------------------------------------ 10 5 50 0.01454808 0.02203783 0.04357100 0.06355273 20 10 200 0.01420087 0.02105476 0.04320518 0.06354146 30 15 450 0.01395945 0.02086954 0.04298302 0.06353145 40 20 800 0.01392084 0.02071138 0.04294779 0.06352954 50 25 1200 0.01388998 0.02068776 0.04291799 0.06352793 60 30 1800 0.01388459 0.02067480 0.04291353 0.06352762 70 35 2400 0.01388310 0.02067227 0.04291193 0.06352753 80 40 3200 0.01388288 0.02067201 0.04291177 0.06352752
Here the convergence is definitely worse than in the two preceding cases.
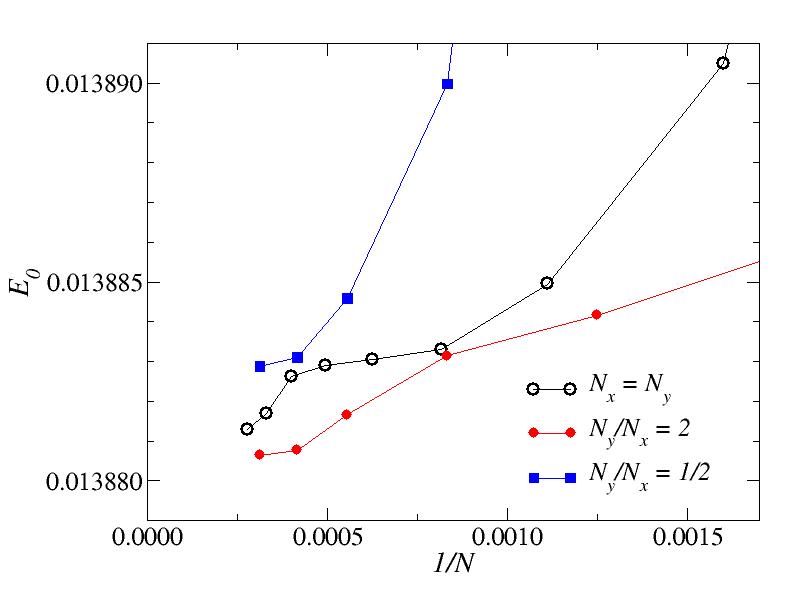
The fiure below shows the data for large N. Regardless of the ratio Ny/Nx, the energy should converge to the same value when N goes to infinity (which seems very plausible here). Note, however, how un-smooth the convergence is. Obviously, not only the shape of the "box" matters in how the energies converge as a function of N and Ny/Nx, but also the internal potential structure. It may not always be easy to see what would be the best ratio to use (and here we have only given a couple of examples, without exploring this in detail).
Now let's look at the wave functions. These were calculated using Nx=Ny=40:
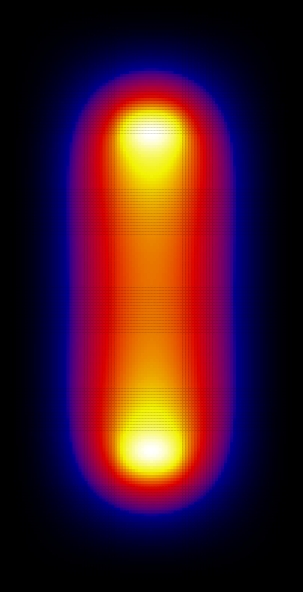
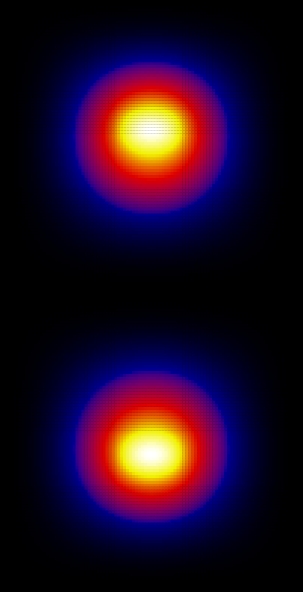
Since the square is graphed we don't see the phase. Looking at the wave function itself, the ground state is (as expected) found to be symmetric and the first excited state is anti-symmetric.
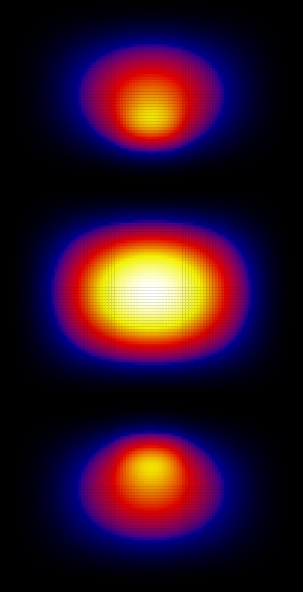
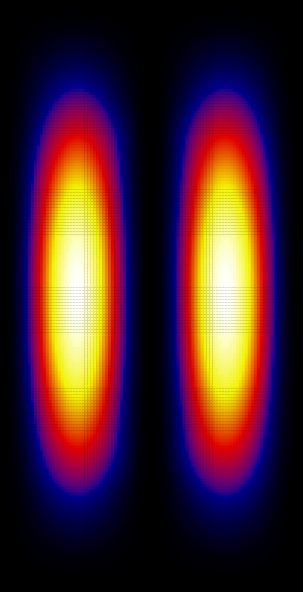
From these graphs we can see that the n=1 and n=3 states look very much like bonding states of a diatomic molecule (large probability between the attractive centers). The n=2 state looks like and anti-bonding state. The n=4 state doesn't really look like a state of a diatomic molecule, because the effects of the finite size of the "box" have become important here, but still the state has some anti-bonding features (low probability in the region between the attractive centers).